








凋亡、自噬与程序性坏死有什么关联吗?
发布时间:2025-12-06
浏览次数:13865
作者:东极药物
细胞死亡是生长和发育中不可缺少的生物学过程,其失调与多种人类疾病的发生密切相关。其中,三种经典的细胞死亡方式——凋亡、自噬和坏死,通过激活特定的信号通路呈现出不同的形态学特征。各种类型的细胞死亡在分子···
细胞死亡是生长和发育中不可缺少的生物学过程,其失调与多种人类疾病的发生密切相关。其中,三种经典的细胞死亡方式——凋亡、自噬和坏死,通过激活特定的信号通路呈现出不同的形态学特征。
各种类型的细胞死亡在分子机制上既有独特性,也存在重叠性。多条信号通路独立调控不同类型的细胞死亡,但它们相互关联、可被同时激活,并能在细胞应激反应中并行运行。
凋亡、坏死与自噬的不同特征
凋亡
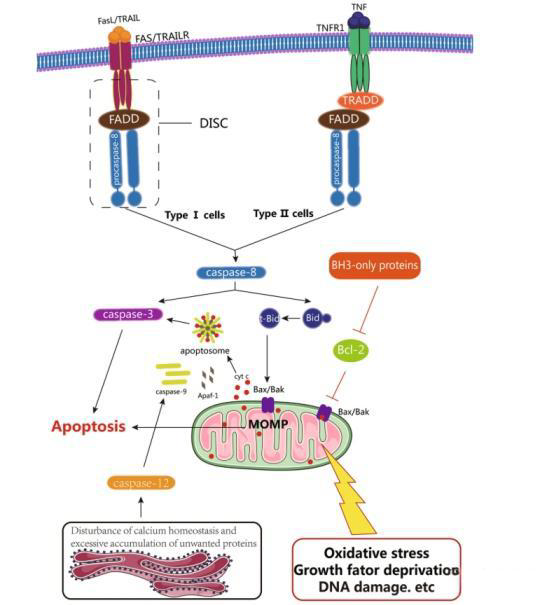
凋亡通常被认为是一种由半胱天冬酶介导的程序性细胞死亡。凋亡细胞呈现出明显的形态学特征,包括细胞体积缩小、染色质凝聚、细胞核碎裂(晚期)、质膜起泡以及凋亡小体的形成;同时伴随生化变化,如磷脂酰-L-丝氨酸在细胞外膜的暴露(早期)。凋亡可通过死亡受体介导的凋亡通路、线粒体依赖性凋亡通路或内质网(ER)应激诱导的凋亡通路被激活。
死亡受体介导的凋亡通路在Fas配体、TNF-α(肿瘤坏死因子α)或TRAIL与相应死亡受体结合后被激活。适配蛋白FADD与procaspase-8形成复合物,即死亡诱导信号复合物(DISC)。在DISC中,procaspase-8通过自水解被激活。活化的caspase-8可通过两种方式传递凋亡信号:一是直接激活caspase-3,二是切割Bid生成截短Bid(tBid)。tBid转移至线粒体,引起Bax和Bak构象变化及其寡聚化,从而在外线粒体膜形成孔道。
线粒体依赖性凋亡通路可被多种应激诱导物激活,如DNA损伤、生长因子缺失及氧化应激。Bcl-2蛋白家族通过调控线粒体外膜通透性控制这一内源性通路。细胞色素c从线粒体释放进入胞质后,与Apaf-1结合,促进caspase-9的激活;随后,caspase-9激活效应性caspase,引发一系列蛋白水解级联反应。
此外,内质网应激(如钙稳态失衡、内质网中过量未折叠或错误折叠蛋白积累、营养缺乏及缺氧)也可诱导凋亡。这类凋亡由caspase-12(一种内质网耐受型半胱天冬酶)介导。活化的caspase-12在从内质网转移至胞质后,可直接切割caspase-9,随后激活caspase-3。ER应激下caspase-12的激活分子机制包括:与肌醇需求酶-1α(IRE1α)-TNF受体相关因子2(TRAF2)复合物形成复合体,或通过钙依赖性胞内半胱氨酸蛋白酶家族calpain介导。
 凋亡示意图
凋亡示意图
自噬
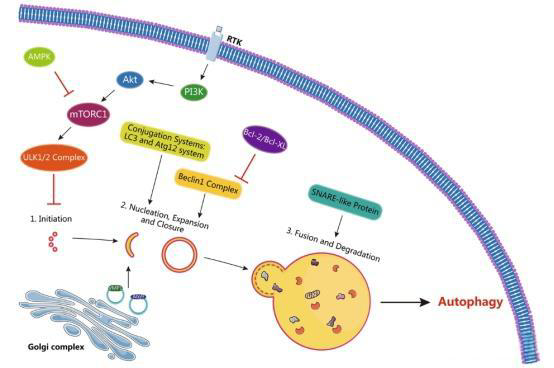
自噬是一种自降解过程,用于应对多种应激,包括营养缺乏、细胞器损伤、缺氧、活性氧(ROS)、内质网应激以及药物处理。自噬过程涉及四个关键步骤——起始、成核、自噬体与溶酶体融合以及水解。
我们对自噬分子机制的理解始于酵母研究。通过酵母的遗传筛选,鉴定出一系列自噬调控分子,尤其是自噬相关蛋白(Atg蛋白),它们是自噬过程的主要执行者。
在起始步骤中,形成吞噬体(phagophore)需要Atg1复合物的组装与聚集,该复合物包括Atg1、Atg13、Atg17、Atg29和Atg31。然而,在哺乳动物中,形成的对应复合物为UNC-51样激酶1(ULK1)-mAtg13-FIP200复合物,包含与酵母Atg1、Atg13和Atg17同源的蛋白。
在成核步骤中,吞噬体在内质网及其他膜上的形成由III类PI-3激酶VPS34、Atg6(哺乳动物中称为Beclin1)、Atg14和Vps15组成的复合物控制。循环于高尔基复合体、自噬体和内体中的Atg9及囊泡膜蛋白VMP1可能参与脂质向隔离膜的运输。
自噬体的扩展与闭合依赖两种泛素样蛋白共轭系统,即Atg12系统和Atg8系统(在哺乳动物中Atg8称为LC3)。Atg12系统包括五种Atg蛋白:Atg5、Atg7、Atg10、Atg12和Atg16。Atg12首先被E1样酶Atg7激活,然后转移至E2样酶Atg10。最终,Atg12的C端甘氨酸共价结合Atg5的Lys149侧链,并与二聚体蛋白Atg16结合形成E3样复合物。
Atg8系统包括四种Atg蛋白:Atg3、Atg4、Atg7和Atg8。Atg8首先被半胱氨酸蛋白酶Atg4切割,暴露其C端甘氨酸(哺乳动物中为LC3 I)。随后,Atg8被E1样酶Atg7激活,并转移至E2样酶Atg3,然后通过E3样Atg12-Atg5-Atg16复合物与磷脂乙醇胺(PE)的氨基共价结合,形成Atg8-PE共价结构(哺乳动物中为LC3 II)。Atg8-PE结构赋予Atg8膜锚定和半融合能力,在自噬体形成中起关键作用。LC3 II存在于自噬体的内外膜上,是自噬形成的典型标志。Atg8-PE共价结构可被Atg4可逆切割为Atg8,实现Atg8的循环利用。
随后,自噬体与溶酶体的融合由SNARE(可溶性N-乙基马来酰亚胺敏感因子结合蛋白受体)类蛋白介导。最终,在低pH环境下,各种溶酶体酶降解所有受损的细胞器、蛋白质、脂质和核酸。
 自噬示意图
自噬示意图
坏死与程序性坏死(Necroptosis)
与凋亡不同,坏死通常被认为是一种非调控、偶发性的细胞死亡。然而,程序性坏死的发现支持了多种非凋亡性调控性细胞死亡机制的存在。目前已报道几种程序性坏死类型,包括程序性坏死(necroptosis)、Parthanatos、铁死亡(ferroptosis)、焦亡(pyroptosis)和网状中性粒细胞外陷阱形成(NETosis)。
其中,程序性坏死,这是一种受调控的坏死性细胞死亡类型,与凋亡共享若干关键信号通路。关于程序性坏死的多数知识来源于对TNF信号的研究。TNF是一种多效性细胞因子,在炎症过程中发挥重要作用。在特定条件下,TNF通过与TNFR结合也能强烈诱导细胞死亡。早期研究显示,TNF可诱导RIPK1介导的半胱天冬酶非依赖性细胞死亡,但这种非凋亡性TNF诱导的细胞死亡并未引起广泛关注,直到研究者进一步发现,当凋亡被阻断时,细胞会执行类似坏死的死亡方式。
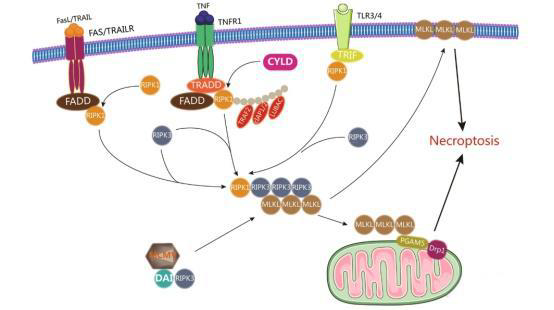
程序性坏死由死亡受体(如TNFR1)和Toll样受体(TLRs)的激活启动。TNF与TNFR结合后,TNFR构象发生变化,招募多种蛋白,包括TNFR1相关死亡结构域蛋白(TRADD)、RIPK1、TRAF2、E3泛素连接酶、cIAP1/2以及LUBAC,形成TRADD和RIPK1依赖的复合物I。该多蛋白复合物通过招募TGF激活激酶1(TAK1)结合蛋白(TAB)复合物及由IKK1、IKK2和NF-κB必需调节因子(NEMO)组成的IκB激酶(IKK)复合物,激活NF-κB信号、AP-1信号及MAPK信号,从而传递促炎和促生存信号。
当RIPK1被环状瘤赖氨酸63去泛素酶(CYLD)去泛素化后,复合物I变得不稳定,RIPK1可解离并形成另一复合物,即复合物IIa,该复合物由TRADD、FADD、procaspase-8和FLIP组成。Procaspase-8与FLIPL一起切割RIPK1,以防止程序性坏死并激活凋亡信号。TNF–TNFR信号还可通过形成复合物IIb诱导凋亡,当IAP(凋亡抑制因子)、TAK1、NEMO和/或Pellino3功能受阻时。复合物IIb包括RIPK1、RIPK3、FADD、procaspase-8和FLIPL,可导致RIPK1依赖性凋亡。然而,当RIPK3和混合谱系激酶样结构蛋白(MLKL)水平足够高且caspase-8活性受抑时,复合物IIb可进一步转化为坏死小体(necrosome),一种微丝样复合物。在坏死小体中,RIPK3的寡聚化和磷酸化促使MLKL被招募并磷酸化,随后MLKL转移至质膜,引起膜损伤并导致程序性坏死。此外,坏死小体可与线粒体膜上的丝氨酸/苏氨酸蛋白磷酸酶PGAM5相互作用,激活线粒体分裂因子动力相关蛋白1(Drp1),通过线粒体碎裂诱导程序性坏死。
如前所述,FasL或TRAIL与死亡受体Fas或TRAILR结合会诱导DISC的形成,激活caspase-8并执行凋亡。然而,在缺乏cIAP或caspase-8受抑时,当Fas/TRAILR被激活时,RIPK1转移至膜上,促进复合物IIb形成并启动程序性坏死。
TLR的激活会形成一个平台,招募胞质适配蛋白TRIF(含Toll/IL-1受体结构域的干扰素-β诱导适配蛋白)。TRIF参与NF-κB信号激活及I型干扰素的诱导。依赖其RHIM(RIP同源相互作用基序)结构,TRIF可与RIPK1和RIPK3相互作用。在凋亡抑制剂zVAD-fmk存在下,脂多糖激活TLR4或聚肌苷-聚胞苷酸激活TLR3可诱导TRIF介导的程序性坏死,该过程可被Necrostatin-1(Nec1)抑制或RIPK1敲除阻断。这表明RIPK1-TRIF信号复合物在TLR3/4诱导的程序性坏死中起重要作用。在缺乏RIPK1的情况下,TRIF也可通过直接招募并激活RIPK3诱导程序性坏死。
除了死亡受体信号和TLR信号,胞质病毒DNA传感器DAI(DNA-dependent activator of IFN-regulatory factors)也可诱导程序性坏死。与TRIF类似,DAI具有RHIM结构。在响应病毒(如小鼠巨细胞病毒)双链DNA时,DAI可激活NF-κB、诱导I型干扰素,并介导RIPK3依赖的程序性坏死。此外,Wei等近期报道了一种新型坏死机制,表明阳离子纳米载体诱导的急性细胞坏死是通过Na+/K+-ATP酶损伤引起的,继而引发炎症反应。
 程序性坏死示意图
程序性坏死示意图
简单来讲:
凋亡是一种由半胱天冬酶介导的程序性细胞死亡,其特点包括染色质凝聚、细胞核碎裂和细胞膜起泡。
与凋亡相反,坏死被认为是一种非调控的、偶发性的细胞死亡,由非特异性或非生理性的应激诱导,其特征包括细胞器膨胀、质膜破裂,以及由细胞内容物释放引发的随后的炎症反应。
自噬则伴随自噬体的形成。自噬体是一种含有受损细胞器、蛋白质及其他胞质成分的双层囊泡。自噬体与溶酶体融合,降解细胞的大分子物质和细胞器,为细胞提供可再生的能量和代谢物。自噬既是一种促生存的机制,也可诱导自噬性细胞死亡。
三种细胞死亡类型之间的联系
凋亡与程序性坏死的关联
凋亡与程序性坏死可能同时发生,或因下游死亡信号通路的相互关联而相互转化。例如,当凋亡被阻断时,细胞可转向坏死性死亡;氧化应激诱导的坏死性细胞死亡也涉及凋亡相关的caspase-8/Bid通路的激活。最终的细胞死亡形式取决于细胞类型、细胞微环境及初始诱导因素。
Caspase-8与RIPK1/3
作为凋亡启动半胱天冬酶,caspase-8与FADD相互作用形成DISC,随后进行同源二聚化和蛋白水解加工。活化的caspase-8随后从DISC释放,触发下游凋亡信号。同时,caspase-8可通过切割并失活RIPK1和RIPK3来抑制程序性坏死。然而,当caspase-8的活性被药理学抑制(如全半胱天冬酶抑制剂ZVAD)或基因干扰抑制如RNAi介导的敲低)时,RIPK1和RIPK3通过磷酸化被激活,并诱导坏死小体形成,从而触发程序性坏死。
ATP
ATP在细胞死亡命运的决策中起关键作用。高水平的细胞内ATP通常有利于凋亡,而低水平则往往促进坏死。因此,细胞内ATP的过度消耗或ATP合成的抑制可能将凋亡转化为坏死。例如,大量DNA损伤可激活聚ADP-核糖聚合酶-1(PARP-1),该核酶参与DNA修复,导致大量NAD+和ATP被消耗,进而引发坏死性死亡。线粒体是ATP的主要生成场所,因此线粒体功能障碍可通过ATP耗竭触发坏死。此外,线粒体过量ROS的形成及线粒体通透性转变(MPT)的发生,也与凋亡向程序性坏死的转化密切相关。
自噬与凋亡的关联
自噬是一种细胞内的分解代谢机制,通过降解和回收胞质中不需要的成分,如功能异常的蛋白质或受损的细胞器,以维持细胞稳态。自噬如同双刃剑,可根据细胞类型、胞内代谢活性、胞外营养供应及触发刺激,既能保护细胞免受凋亡,也可促进凋亡的发生。
Beclin1
哺乳动物Beclin1(酵母中为Atg6)通过与抗凋亡蛋白家族的直接相互作用,实现对自噬和凋亡的交叉调控。Beclin1是自噬体形成的关键分子。Beclin1可与III类PI3K复合物PI3KC3/Vps34相互作用,促进Beclin1-Vps34-Vps15核心复合物的形成。Beclin1还是BH3-only蛋白家族的成员。抗凋亡蛋白Bcl-2或Bcl-xL可通过BH3结构域与Beclin1结合,同时抑制Beclin1活性,从而阻断自噬过程,并抑制内源性凋亡的发生。另一方面,NOXA及其他BH3-only家族蛋白可将Bcl-2家族蛋白从Beclin1中置换出来,促进自噬性细胞死亡。此外,Beclin1也可被多种半胱天冬酶切割,如caspase-8和caspase-3,使细胞命运从自噬转向凋亡。Beclin1的C端片段随后可转移至线粒体,诱导线粒体膜通透性增加并触发凋亡。
自噬与程序性坏死的关联
自噬与程序性坏死之间的相互关系已在多项研究中被探讨,但结果存在一定争议。作为一种保护性机制,自噬可以抑制程序性坏死,这并不意外。有趣的是,在某些情况下,自噬似乎反而促进程序性坏死。例如,Khan等报道,棕榈酸可触发Ca²⁺依赖性自噬,导致内皮细胞发生程序性坏死。
mTORC1
mTORC1是感知营养、成长因子及应激的关键传感器,控制细胞代谢、生长和存活。生长因子和营养激活mTORC1后,可通过磷酸化参与自噬起始的自噬相关蛋白(如ULK复合物中的ULK1和ATG13,以及VPS34复合物中的ATG14)抑制自噬。细胞的代谢和能量状态可调控mTORC1活性,从而影响自噬的诱导。在低能量状态下,AMP/ATP比值升高会激活AMPK信号,AMPK通过磷酸化TSC2(一种mTORC1负调控因子)抑制mTORC1信号通路,进而促进自噬。
通过下调mTORC1信号刺激自噬,可在营养或能量缺乏的条件下保护细胞免受程序性细胞死亡(包括凋亡和程序性坏死)的侵害。如前所述,DNA损伤反应中PARP-1的激活会因ATP耗竭导致坏死,同时也会激活AMPK、抑制mTORC1并诱导自噬作为最后的保护手段。自噬与坏死之间的平衡最终决定细胞死亡的命运。
凋亡、自噬与程序性坏死的相互关联
细胞FILCE样抑制蛋白(cFLIP)
FADD样白介素-1β转换酶(FLICE)样抑制蛋白(FLIP)具有类似caspase-8的结构,但缺乏蛋白水解活性。人类cFLIP主要有三种异构体:一种长蛋白cFLIPL和两种短蛋白cFLIPs及cFLIPR。cFLIPL具有C端caspase-8样结构域,但由于若干关键催化氨基酸残基被替换,失去了酶活性;而短异构体cFLIPs和cFLIPR则不具备caspase样C端结构域。然而,这三种异构体的N端均含有两个死亡受体结构域,使其能够与适配蛋白FADD相互作用,形成DISC复合物。
cFLIP不仅调控死亡受体介导的外源性凋亡通路,还可调控死亡受体非依赖的凋亡通路。在复合物IIb/程序性凋亡体(ripoptosome)中,同源二聚的caspase-8通过切割RIPK1并解体复合物IIb/ripoptosome来启动凋亡。当procaspase-8与cFLIPL形成异源二聚体时,由于RIPK1被切割,程序性坏死被阻止;同时,活化caspase-8未形成,因此凋亡也被阻断。然而,当procaspase-8与cFLIPs或cFLIPR形成异源二聚体时,由于RIPK1缺乏蛋白水解切割,程序性坏死被触发,同时无法诱导caspase-8依赖性凋亡。因此,ripoptosome中cFLIP异构体的存在决定了细胞是执行RIPK1依赖的程序性坏死,还是caspase-8依赖的凋亡。
除了调控凋亡和程序性坏死外,cFLIP还是自噬的负调控因子。在自噬体形成过程中,Atg3共价结合微管相关蛋白LC3。令人注意的是,cFLIP通过竞争性结合Atg3,阻止Atg3与LC3的结合,从而抑制自噬。cFLIP通过ripoptosome调控凋亡和程序性坏死的过程发生在质膜上,而其抑制自噬的作用则发生在自噬体形成部位。因此,cFLIP在不同亚细胞定位可能对其功能具有重要意义。
文章出自:验外实包 想了解更多请关注:http://www.dj-cro.com/
上一篇:免疫沉淀实验
下一篇:化学方法建立的慢性肾功能衰竭模型
本文标签:












